The near-complete genome assembly of Reynoutria multiflora reveals the genetic basis of stilbene and anthraquinone biosynthesis
何首乌几乎完整的基因组组装揭示了芪类化合物和蒽醌类化合物生物合成的遗传基础

摘要
何首乌(Reynoutria multiflora)是中国广泛使用的药用植物。其药用成分主要是芪类化合物(stilbenes)和蒽醌类化合物(anthraquinones),具有抗衰老、抗炎和抗氧化等重要药理活性,但其生物合成途径仍未完全阐明。在本研究中,我们报道了何首乌的近乎完整的基因组组装,大小为1.39 Gb,contig N50为122.91 Mb,仅剩一个空隙。基因组进化分析表明,两个最近的长末端重复(LTR)爆发显著推动了何首乌基因组大小的增加,并且在何首乌和荞麦基因组之间观察到大量的大规模染色体重排。比较基因组学分析揭示,属于蓼科的全基因组复制事件显著扩展了与抗病性和芪类及蒽醌类化合物生物合成相关的基因家族。结合转录组学和代谢组学分析,我们阐明了不同生长年份何首乌根部药用成分含量动态变化的分子机制。此外,我们还鉴定了几种可能与蒽醌和芪类化合物生物合成相关的关键基因。我们发现一个在根部高表达的芪类合成酶基因PM0G05131,它可能在何首乌芪类化合物的积累中发挥重要作用。这些基因组数据将加速揭示药用植物中蒽醌和芪类化合物的生物合成途径。
1 引言
何首乌(Reynoutria multiflora),属于蓼科,是一种广泛应用的传统中草药。何首乌起源于中国,具有重要的药用价值,已被广泛应用于亚洲、欧洲和美洲的多种草药配方、膳食补充剂和天然药物中(Sun等,2013;Kim & Kim,2018)。何首乌的年消费量超过10,000吨,已成为中国草药产业链中的重要组成部分(Han等,2021)。现代药理研究表明,何首乌根部富含芪类化合物和蒽醌类化合物,具有黑发、抗衰老、抗高脂血症、抗氧化和抗癌等多种作用(Chan等,2002;Yang,2005;Steele等,2013;Lin等,2015)。根据《中国药典》,2,3,5,4′-四羟基芪类-2-O-ß-D-葡萄糖苷(THSG)属于芪类化合物,蒽醌类的主要成分包括大黄素(emodin)和大黄素醇(physcion),被认为是评估何首乌药材质量的重要标志物(Lv等,2007;Lin等,2015)。尽管何首乌在药物和保健品中的应用广泛,但其丰富的药用活性成分的遗传基础尚未被深入研究,主要是因为缺乏高质量的参考基因组(Zhao等,2014)。
2,3,5,4′-四羟基芪类-2-O-ß-D-葡萄糖苷、大黄素和大黄素醇被认为是何首乌中最关键的药用化合物(Wang,M等,2012)。之前的研究表明,THSG、大黄素和大黄素醇具有共同的前体——马洛酰辅酶A(malonyl Co-A),并通过糖基化稳定地存在于植物中(Xia等,2018;Yin等,2020;Kang,S-H等,2021)。白藜芦醇(3,5,4′-三羟基芪类)是最为研究的芪类化合物,通过苯丙氨酸生物合成途径合成,包括四种主要酶:苯丙氨酸氨基裂解酶(PAL)、肉桂酸4-羟化酶(C4H)、4-香豆酰辅酶A连接酶(4CL)和芪类合成酶(STS)(Zhong等,2004;Zhao等,2014;Zhang等,2019)。在这些酶中,STS是一种植物特异性的III型聚酮合成酶(PKS),可以催化一个4-香豆酰辅酶A分子和三个马洛酰辅酶A分子缩合,合成白藜芦醇(Watanabe等,2007)。与白藜芦醇相比,2,3,5,4′-四羟基芪类-2-O-ß-D-葡萄糖苷在C2位置多了一个O-葡萄糖苷,之前的研究表明,何首乌中一个芪类合成酶基因RmPKS和若干CYP450基因与不同组织中THSG的积累密切相关(Sheng等,2010)。然而,从白藜芦醇到THSG的生物合成途径以及相关的关键酶仍然没有得到充分阐明。
近年来的研究推测,植物中蒽醌类化合物的生物合成主要通过聚酮途径(Karppinen等,2008;Mizuuchi等,2009;Kang等,2021)。在聚酮途径中,CHS是蒽醌生物合成中的首个限速酶,已被报道能够有效催化马洛酰辅酶A的七次脱羧缩合反应,生成八聚酮链(Kang等,2021)。聚酮环化酶(PKCs)被认为催化八聚酮形成芳香环,从而生成蒽醌中间体(Iyer等,2001),并进一步经过脱羧和脱水反应生成大黄素蒽酮,再经过氧化生成大黄素(Karppinen等,2008;Mizuuchi等,2009;Kang等,2021)。一个甲基基团被添加到大黄素的C3位置生成大黄素醇(Kang等,2021),O-甲基转移酶(OMT)——一类催化甲基转移的酶(Parajuli等,2018)推测在大黄素醇的生成中发挥作用。通常,糖基化是通过UDP-葡萄糖转移酶(UGT)在药用植物中生成糖苷的主要机制,大大提高其稳定性和溶解性,从而决定其药理功能(Lv等,2007;Zhang,D等,2017)。UDP-葡萄糖转移酶在植物次级代谢中起着重要作用,具有广泛的底物特异性,例如在荞麦中,FtUGT73BE5被证明在体外负责大黄素的糖基化(Yin等,2020)。尽管已有很多与蒽醌生物合成相关的酶被报道,但何首乌中丰富的蒽醌类化合物的遗传基础仍然不清楚。
已有研究表明,药用植物根茎中有效成分的积累与生长年份相关(Bai等,2020)。例如,研究表明,党参中人参皂苷的含量随着时间的推移不断增加(Zhang等,2017),但某些药用植物的几种活性成分的积累与生长年份呈负相关,如远志中的皂苷(Teng等,2009)。因此,必须综合评估根茎药用植物的生物量和药用成分,以确定最佳的采收时间。之前的研究表明,何首乌根部的THSG、大黄素和大黄素醇含量随着生长年份的增加而提高(Zhao等,2021)。然而,何首乌中这些活性成分的动态变化及其背后的遗传机制仍然不清楚。
近年来,随着测序技术的发展,针对许多药用植物的基因组和多组学分析已开展,以鉴定催化植物活性物质生物合成的关键酶基因,并阐明其分子机制。在本研究中,我们提供了何首乌近乎完整的染色体级基因组组装,研究了与蒽醌和芪类化合物生物合成相关的候选基因家族的进化,并揭示了何首乌中丰富的药用活性成分的遗传基础。结合基因组学、转录组学和代谢组学分析,我们系统推测了THSG、大黄素和大黄素醇的生物合成途径,揭示了何首乌根部活性成分在生长过程中动态变化的分子机制,并鉴定了几个与蒽醌和芪类化合物生物合成相关的基因。这些基因组数据将为指导及时采收并帮助发现与何首乌中活性成分合成相关的功能基因提供基础资源。
2 材料与方法
2.1 植物材料与基因组大小评估
我们在2021年从山东省青州市(118°E,36°N)采集了相同品种的何首乌(Reynoutria multiflora),并在河北大学生命科学学院的温室内维持该植物,温度为25–30°C,每天提供14小时的光照。然后,通过块茎大小确定了何首乌的生长年限,例如:1年生(0.91.2 cm)、3年生(2.63.0 cm)、5年生(6.57.2 cm)和10年生(1012 cm)。最后,选择了1年、3年、5年和10年生的块茎用于代谢组学和转录组学测序。通过使用DNA Safe Plant Kit(Tiangen,中国)从5年生的叶片中提取高质量基因组DNA。与此同时,从5年生的植物中采集了三种植物组织(根、茎、叶),并将其冻存于液氮中,以便进行RNA提取和RNA-seq,三个生物学重复。使用RNeasy Plant Mini Kit(Qiagen,美国)从收集的样本中提取总RNA。为了测量何首乌的核DNA含量,使用番茄和玉米作为内控,通过流式细胞术根据荧光强度和基因组大小的相等比例进行比较。根据先前描述的方法(Doležel等,2007),通过与番茄和玉米的荧光强度比较来估算何首乌的基因组大小。
2.2 基因组测序
对于短读序列测序,构建了插入大小为350 bp的Illumina文库,并使用Illumina平台进行测序。对于PacBio SMRT测序,使用SMRTbell Express模板准备套件2.0构建了两个(15–20 kb)的SMRT Bell文库,并使用单分子实时(SMRT)测序技术在PacBio Sequel II平台上进行测序。
Hi-C文库的构建和测序由Berry Genomics公司进行。简而言之,首先对5年生的何首乌嫩叶进行固定和裂解,之后使用MboI酶对交联的DNA进行过夜消化。酶被灭活,粘性末端通过添加生物素标记的dCTP进行填充。在钝端连接缓冲液中进行接近连接后,逆转交联并纯化DNA以构建Hi-C文库。最终文库在Illumina HiSeq 2500平台上以150-bp双端模式进行测序。
2.3 基因组组装与评估
何首乌基因组的基因组大小和杂合度是基于流式细胞术和k-mer频率分析(使用GCE软件v1.0.2)确定的,使用默认参数进行17-K-mer分布分析。然后,使用来自PacBio SMRT、Hi-C和Illumina PE150的测序数据,获得何首乌的高质量基因组序列。所有由PacBio SMRT测序仪产生的HiFi读段数据都用于使用Hifisam(v0.16.1)进行基因组组装(Cheng等,2021),并使用默认参数。为了将scaffold定位到染色体上,高通量染色体构象捕获(Hi-C)测序数据通过Juicer v1.5(Durand等,2016)和3D-DNA(Dudchenko等,2017)进行了比对。通过Hi-C图谱和比对信息,使用Juicebox手动检查和修正潜在的组装错误。组装质量通过使用BWA(v0.7.17)(Li & Durbin,2009)将Illumina平台的短读序列比对到基因组上进行评估。此外,使用LTR_retriever(v2.9.0)(Ou等,2018)进行LAI(LTR组装指数)分析,并使用BUSCO(v5.2.2,基准通用单拷贝同源基因)分析来评估组装的完整性。
2.4 基因组注释
结合两种方法,采用基于同源性搜索和自定义方法识别何首乌基因组中的重复序列。对于自定义方法,使用RepeatModeler(v1.0.11)(RepeatModeler Open-1.0)、LTR_FINDER(v1.07)(Xu & Wang,2007)、LTRharvest(Ellinghaus等,2008)和LTR_retriever(v2.9.0)(Ou & Jiang,2018)构建基于de novo的重复库,使用默认参数。使用RepeatMasker(v4.1.1)(Tarailo-Graovac & Chen,2009)识别重复序列。对于同源性搜索,使用已知的重复库(Repbase v15.02)通过RepeatMasker识别重复元素。我们使用INFERNAL(Nawrocki等,2009)软件,基于Rfam数据库(9.1版本)预测tRNA、rRNA、miRNA和snRNA。
使用三种方法预测何首乌中的蛋白编码基因。首先,使用来自RNA-Seq数据集的高质量蛋白质作为训练集,对Augustus(v3.2.3)(Stanke & Waack,2003)进行基因预测。对于基于同源的预测,使用六种物种的蛋白质序列,包括拟南芥(Arabidopsis thaliana)、荞麦(F. tataricum)、甜菜(Beta vulgaris)、土豆(Solanum tuberosum)、番茄(Solanum lycopersicum)和仙人掌(Hylocereus undulatus),使用Genomethreader(v1.7.3)(Gremme等,2005)对基因组进行搜索。对于基于RNA-seq的预测,使用Trinity(v2.12.0)(Haas等,2013)进行RNA-seq数据的组装,然后使用PASA软件;RNA-seq数据也通过HISAT2(v2.2.1)(Kim等,2019)与基因组比对,并使用StringTie(v2.1.6)(Pertea等,2015)将比对的读段组装成转录本,并使用TransDecoder(v5.1.0)预测最长的开放阅读框(ORF)。最后,使用EVidenceModeler(Haas等,2008)将三种方法的基因模型进行整合,生成非冗余的基因集。使用BUSCO评估基因模型的完整性和质量。然后,通过blast搜索对蛋白编码基因进行功能注释,E值的截止值为1e−5,包括Interproscan(v5.32)、UniProtKB/Swiss-Prot和NCBI数据库。通过InterProScan进行Gene Ontology(GO)注释和Pfam域注释。通过同源搜索KofamScan(Aramaki等,2020)进行KEGG同源注释(KO)术语的分配。
2.5 比较基因组分析
通过OrthoFinder(v2.3.8)(Emms & Kelly,2019)使用默认参数将何首乌和其他15个植物物种(F. tataricum、Rheum nobile、Fagopyrum dibotrys、Fagopyrum esculentum、Spinacia oleracea、B. vulgaris、Hylocereus undatus、Simmondsia chinensis、A. thaliana、Theobroma cacao、Populus trichocarpa、Glycine max、Vitis vinifera、Oryza sativa和Zea mays)的蛋白质序列聚类为同源群体。通过识别单拷贝基因,构建系统发育树,使用RAxML(v8.2.12)(Stamatakis,2014)与PROTGAMMAJTT模型和100个bootstrap重复进行。利用TimeTree数据库的三个修正的分歧时间点(27.9–62.4百万年前[S. oleracea与B. vulgaris之间]、89–97.4百万年前[T. cacao与A. thaliana之间]、41.4–51.9百万年前[O. sativa与Z. mays之间]),使用r8s程序估算15个物种的分歧时间。通过比较祖先和每个物种之间聚类大小的差异,使用CAFÉ(v4.2.1)程序(Han等,2013)确定同源基因家族的扩展和收缩。通过R包clusterProfiler(Yu等,2012)进行GO和KEGG功能富集分析。我们使用jcvi(v1.1.19,MCscan for python)(Wang,Y等,2012)来识别结构变异。为了研究全基因组复制(WGD)事件,我们使用McscanX识别基于每个物种内所有对比的局部BLASTP(E值 < 1 × 10−5)的共线性区块,并计算共线性区块基因的同义替代率(Ks),使用KaKs-Calculator(v2.0)和YN模型。然后,使用MCScanX包中的“duplicate_gene_classifier”程序对四种类型的重复基因进行分类,包括全基因组或片段重复、串联重复、近端重复或分散重复。使用iTAK(Zheng等,2016)和RGAugury(Li等,2016)分别基于基因组序列鉴定转录因子基因和抗逆基因。通过R包clusterProfiler(Yu等,2012)进行GO和KEGG功能富集分析。
HiC-Pro(v3.1.0)(Servant等,2015)和Cworld compartments被用于在何首乌和荞麦的每个染色体500-kb修正矩阵上识别A和B区。对于每个染色体,通过第一特征向量(PC1)正值或负值为区分A或B区。PC1方向相同、基因数目更多、表达水平较高的区域对应于A区,而PC1方向相反的区域则属于B区。
2.6 转录组分析与共表达分析
原始RNA-seq读数数据通过去除接头序列、去除含N大于10%和低质量碱基(Q < 20)占比超过50%的读段进行过滤。随后,使用HISAT2软件将清洗后的读段比对到参考基因组。基于每千碱基转录本每百万片段映射的片段数(FPKM),使用StringTie计算基因的表达水平。使用EdgeR检测差异表达基因(DEGs),标准为|log2Fold Change| ≥ 1和假发现率(FDR)< 0.05。使用R包对DEGs进行聚类分析,并对每个聚类中的基因进行功能富集分析。
在共表达网络的构建中,我们使用了上述四个样本(1年生、3年生、5年生和10年生的根)中的差异基因,并要求在至少一个样本中FPKM ≥ 0.1的基因被纳入分析。为了分析代谢物和差异基因之间的网络调控,分别处理了代谢物积累数据和基因表达数据。通过相关性网络分析建立了基因与代谢物的共调控模式。我们使用R包WGCNA(Langfelder & Horvath,2009)进行了加权基因共表达分析。使用软阈值功率为30和默认参数构建了签名共表达网络。控制共表达模块之间最小距离的mergeCutHeight参数设置为0.25。最终,我们获得了与药用成分(如芪类和蒽醌类化合物)调控密切相关的基因模块。通过模块与各种代谢物的相关系数提取与代谢物相关性高的模块基因,并使用Cytoscape v3.9.1(Shannon等,2003)可视化网络。
为了识别与生长过程中核心酶基因相关的转录因子,我们使用了一种重建时间排序基因共表达网络(TO-GCNs)的新方法(Chang等,2019)。通过Pearson相关性(>0.95)确定981个转录因子(TFs)与14,524个基因在GCN中的时间排序(级别)。我们利用TO-GCN预测了STS基因的直接调控候选因子,这些候选因子应与特定结构基因在相同水平或较早的水平共表达。此外,提取了五个STS基因上游2kb的序列,以识别潜在的转录因子结合位点及上游调控路径。然后,使用PlantCARE和PlantRegMap预测了疑似启动子序列的转录因子结合位点,P值 ≤ 1e−4,q值 ≤ 0.05。最后,使用R包drawProteins展示了通过PlantRegMap揭示的转录因子结合位点。
2.7 代谢组分析
准备了来自不同年份的根样本(三个生物学重复),并根据Metware Biotechnology Co., Ltd.(武汉,中国)提供的方法提取代谢物(Tang等,2021;Yuan等,2022)。简而言之,将冻干样本在混合磨机(MM 400;德国Retsch)中以30Hz频率研磨1.5分钟。然后,取50mg冻干粉末,溶解于1.2mL 70%甲醇提取液中,每30分钟涡旋30秒,共涡旋六次。之后,将样品在4°C下以12,000r/min离心3分钟,使用微孔膜(SCAA-104,0.22mm孔径;ANPEL,上海,中国)过滤提取物。
使用超高效液相色谱-质谱联用系统(UPLC-MS)分析样品提取物(Chen等,2013)。分析条件如下:高效液相色谱柱:shim-pack VP-ODS C18(孔径5.0μm,长2×150mm);溶剂系统:水(0.04%醋酸):乙腈(0.04%醋酸);梯度程序:0分钟时为100:0 V/V,20.0分钟时为5:95 V/V,22.0分钟时为5:95 V/V,22.1分钟时为95:5 V/V,28.0分钟时为95:5 V/V;流速:0.25mL/min;温度:40°C;进样量:5μL。流出液交替连接到ESI三重四极杆线性离子阱(QTRAP)质谱仪。ESI源操作参数如下:源温度550°C;离子喷雾电压(IS)5500V(正离子模式)/−4500V(负离子模式);离子源气体I(GSI)、气体II(GSII)、帷幕气(CUR)设置为50、60和25 psi;碰撞激活解离(CAD)设置为高。仪器调谐和质量标定分别使用10μmol/L和100μmol/L的聚丙烯醇溶液在QQQ和LIT模式下进行。QQQ扫描以多反应监测(MRM)实验进行,碰撞气体(氮气)设置为中等。每个MRM过渡的解聚电势(DP)和碰撞能量(CE)通过进一步优化来调整。针对代谢物的不同保留时间,监测特定的MRM过渡。最后,制备质量控制样品(混合物),将所有样品等量混合,以监测分析条件的稳定性。使用UPLC-ESI-MS/MS系统检测代谢物谱。所有代谢物的定性分析根据自建的Metware数据库(Metware Biotechnology Co., Ltd.,武汉,中国)和其他公共代谢物数据库,包括MassBank、KNAPSAcK、HMDB和METLIN(Zhu等,2013;Yuan等,2022)。质谱数据随后使用Analyst(v1.6.3)和Multiquant(v3.0.2)软件进行分析和定量。
2.8 蒽醌类和THSG生物合成途径的基因鉴定
通过以前的研究以及从NCBI和Uniprot数据库中获取了参与生物合成途径的关键酶编码基因。这些序列通过BLASTP(E值<1e−5)与R. multiflora基因组比对,结果筛选出相似度>40%和覆盖率>50%的基因(Li等,2021)。通过PFAM数据库(E值<1e−5)进一步确认最终的候选酶基因。
2.9 CYP450和UGTs基因家族鉴定
通过使用HMMER(v3.0)(Eddy,2009)对相应的Pfam家族PF00067(P450)和PF00201(UGT)域进行HMM轮廓比对,鉴定了CYP450和UGT家族成员。在去除编码小于200个氨基酸的蛋白质序列后(Cheng等,2020),共有276个CYP450和126个UGT进行进一步分析。候选序列通过保守域搜索数据库(CDD:NCBI Conserved Domain Search)进一步确认。使用MEGA6软件中的邻接法构建了CYP450和UGT的系统发育树,使用1000次bootstrap重复。
3 结果
3.1 R. multiflora的高质量基因组组装与注释
何首乌(Reynoutria multiflora)为二倍体(2n = 2x = 22;图1A,B),根据k-mer分析,估算的基因组大小为1.40 Gb,杂合度为0.87%(图S1),流式细胞术测得的基因组大小为1.35 Gb(图S2)。为了获得高质量基因组,我们产生了61.94 Gb的PacBio HiFi读取数据和121 Gb的Hi-C配对读取数据(表S1)。最终组装的基因组总长度为1.39 Gb,contig N50为122.91 Mb,组装的基因组大小与k-mer分析和流式细胞术估算的基因组大小相似(表S2)。R. multiflora的基因组质量(contig N50 122.91 Mb)显著高于最近发布的草本植物基因组组装,包括大青(contig N50 1.47 Mb)(Kang等,2021)、虎杖(contig N50 4.21 Mb)(Wang等,2021)、木贼(contig N50 4.03 Mb)(Kang等,2021)和黄连(contig N50 806.60 kb)(Liu等,2021)。使用Hi-C接触信息,进一步将这些contig锚定到11个假染色体上,只有一个空隙,覆盖了约99.87%的组装序列(图1C, 1D;表S3)。假染色体的长度从112.06 Mb到140.70 Mb不等,平均长度为126.73 Mb(表S3)。值得注意的是,10个假染色体由单个contig组成。17个端粒区域在11个染色体中被鉴定出,其中7个为端粒到端粒的染色体(表S4)。BUSCO结果显示,约97.2%的完全的被子植物基因在组装基因组中被识别(表S5)。LAI得分为17.43(图S3和表S6),达到参考质量标准(Ou & Jiang,2018)。Illumina DNA短读和RNA-seq读取的比对率分别为约99.61%和94.78%(表S7)。这些结果表明,我们提供了一个近乎完整的R. multiflora基因组,具有较高的连续性、完整性和准确性。
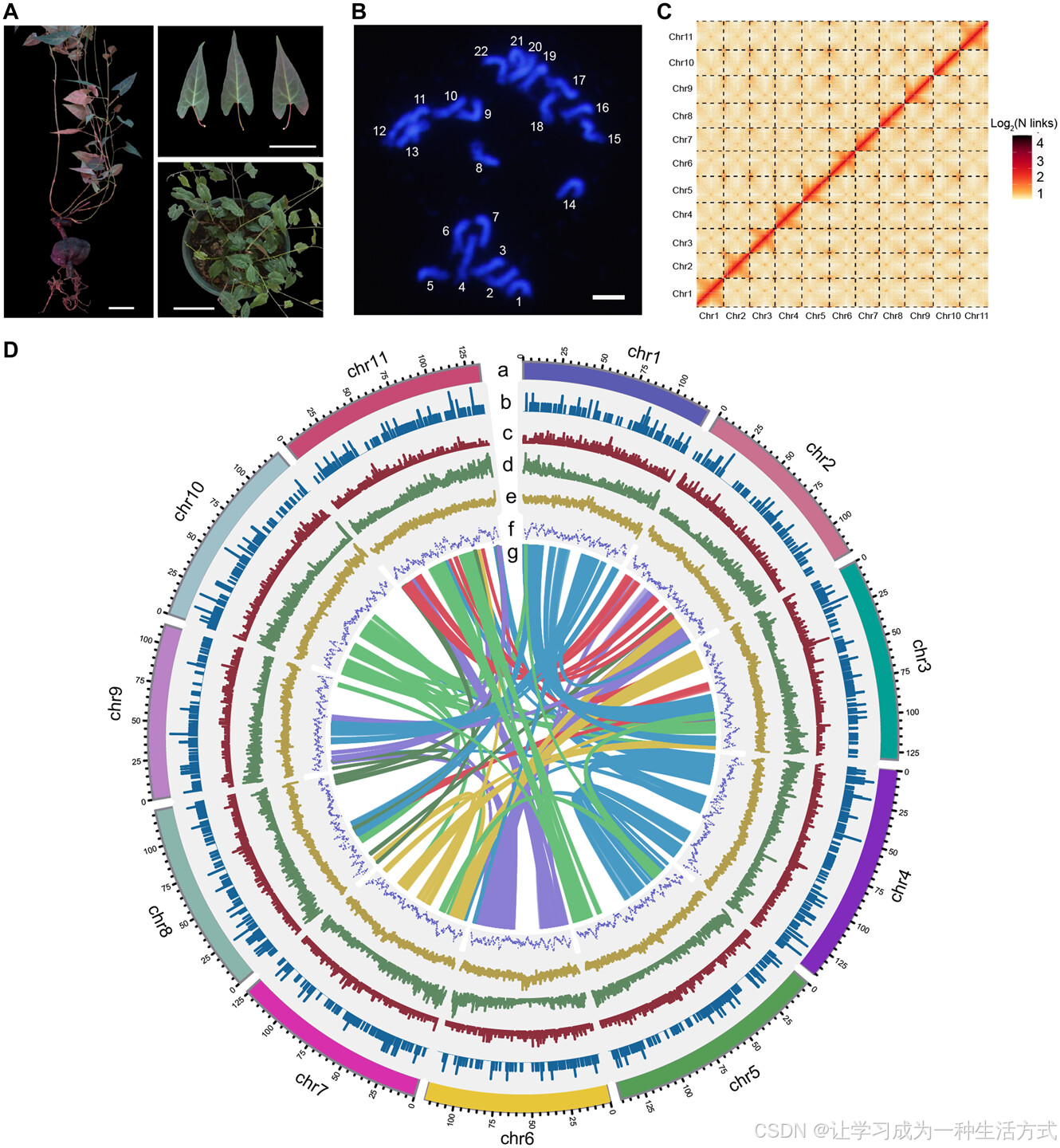
何首乌的表型、核型和基因组特征
A,何首乌(R. multiflora)叶片、茎和根的形态(比例尺 = 5 cm)。 B,使用DAPI染色的何首乌染色体的荧光图像(2n = 2x = 22)。 C,Hi-C染色体相互作用的基因组强度信号热图。 D,何首乌基因组特征的分布。轨道“a”表示组装的染色体,轨道“b–f”分别表示LTR Copia密度、LTR Gypsy密度、基因密度、GC含量和LAI。每条连接线位于圆圈的中心,连接轨道“g”中同源基因对。颜色表示染色体上每200-kb滑动窗口中的基因组特征密度。
大约921 Mb(66.12%)的何首乌基因组被注释为重复序列(表1,S8)。其中,长末端重复逆转录转座子(LTR-RTs)是主要成分,占基因组的50.48%。其中,Gypsy元素(41.86%)是最丰富的LTR-RT类型,其次是Copia元素(7.83%)。此外,鉴定出16,204个全长LTR-RT元素,包括8416个Gypsy和1222个Copia元素(表S9)。预测了总共42,529个基因模型,平均CDS长度为1059 bp,平均外显子数为4.7(表S10)。98%的被子植物BUSCO基因在何首乌基因集中被鉴定出(表S11),表明基因注释的完整性。何首乌的CDS、外显子和内含子长度分布与相关物种相似(表S12),并且86.5%的编码基因通过Interproscan、Uniprot和KEGG数据库进行了功能注释,证明了我们基因注释的高质量。此外,我们还鉴定了非编码RNA(ncRNA)基因,包括8078个rRNA、4996个tRNA、323个miRNA和4199个snRNA基因(表S13)。
Table 1. Statistics of genome assembly
| Reynoutria multiflora | |
|---|---|
| Genome assembly | |
| Estimated the genome size (Gb) | 1.40 |
| Assembled genome size (Gb) | 1.39 |
| Number of contigs | 19 |
| Contig N50 (Mb) | 122.91 |
| Longest scaffold (Mb) | 140.70 |
| Chromosome numbers | 11 |
| Anchored rate (%) | 99.87 |
| GC content | 38.87% |
| BUSCO | 97.2% |
| LAI | 17.43 |
| Genome assembly annotation | |
| Repeat region % of assembly | 66.12% |
| Predicted gene models | 42 529 |
| Average coding sequence length (bp) | 1056 |
| Average exons per gene | 4.6 |
| Average exon length (bp) | 231 |
-
LAI, LTR Assembly Index.
3.2 比较基因组学分析揭示了何首乌(R. multiflora)与荞麦(F. tataricum)基因组之间的广泛重排
为了研究何首乌的进化,我们构建了一个系统发育树,并使用113个单拷贝基因估算了15个植物物种的分歧时间(图2A;表S14)。主干和各分支之间的系统发育关系与先前的研究结果一致(图2A)(Zhang等,2022;He等,2023;Li等,2023)。分子定年分析显示,何首乌与紫花苜蓿(R. nobile)为姐妹群体,两者的分歧发生在大约17.17百万年前(图2A)。我们发现,何首乌的基因组大小(1.39 Gb)明显大于荞麦的基因组(0.49 Gb)。跨基因组比较揭示,何首乌中的LTR-RTs,特别是Gypsy元素,经历了两次近期的大规模爆发,分别出现在约0.4百万年和1.1百万年左右(图2B、2C、S4),这直接导致了何首乌基因组大小显著增加。具体而言,何首乌的LTR-RTs比荞麦多约697 Mb,占何首乌基因组相对于荞麦增加的936 Mb的74.47%。
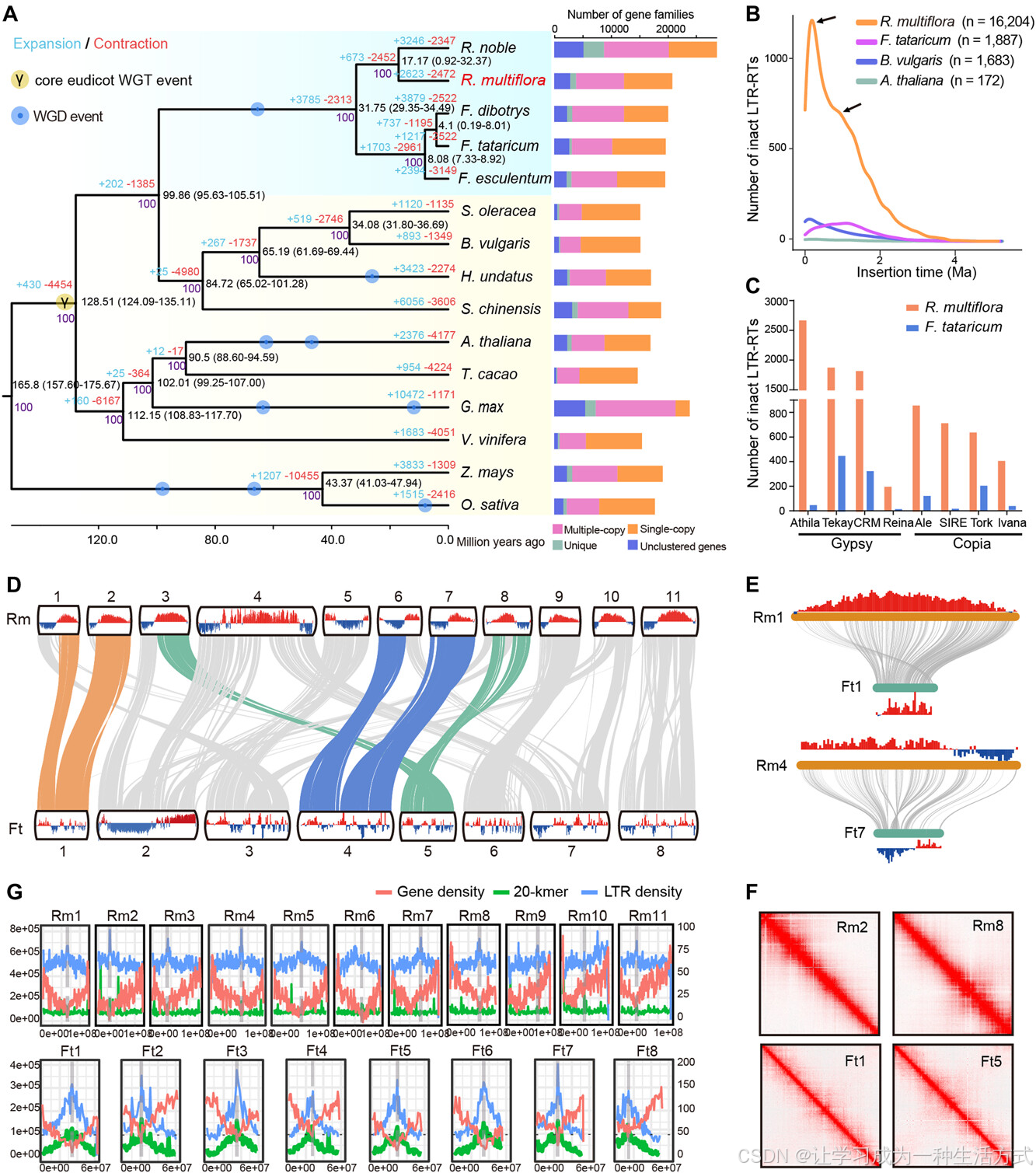
A,基于来自15个植物物种的113个单拷贝同源基因构建的系统发育树,右侧面板展示了每个物种中基因家族的分布。黑色数字表示每个节点的分歧时间(单位:百万年前),扩展和收缩的基因家族数目分别用蓝色和红色标出。 B,何首乌(R. multiflora,简称Rm)、荞麦(F. tataricum,简称Ft)、拟南芥(A. thaliana)和甜菜(B. vulgaris)中LTR-RT插入爆发的时间模式。每个物种使用的完整LTR-RT的数量在括号中提供。黑色箭头表示两次LTR扩展。 C,四个Gypsy LTR家族(Athila、Tekay、CRM和Reina)和四个Copia LTR家族(Ale、SIRE、Tork和Ivana)在何首乌中相对于荞麦的丰度增加。 D,何首乌(Rm)与荞麦(Ft)基因组的基因组共线性分析。染色体之间的共线性区块用灰线表示,区块上下部分分别表示A/B compartment的状态。红色表示A compartment,蓝色表示B compartment。 E,何首乌(Rm)中的Fm1和Fm4染色体的共线性区块与荞麦中的相应区域显示出保守的A compartment区域和交换的A/B compartment区域。 F,使用Hi-C热图验证了大片段重排。 G,候选着丝粒区域的鉴定。红色、绿色和蓝色线条分别表示基因密度、20-mer频率和LTR密度。灰色虚线表示候选着丝粒。
何首乌(R. multiflora)的基本染色体数(11)与荞麦(F. tataricum,8)不同,但在整个染色体层面上,两者的基因组展现出了高共线性,通过比对两种基因组,94.64%的共线性区域得到了确认(图2D)。与此同时,在何首乌与荞麦的基因组中观察到了大量的基因组重排事件,只有两个染色体呈现几乎1:1的共线性关系(图2D)。这些重排通过Hi-C热图进一步得到了验证(图2F)。有趣的是,何首乌和荞麦中的大多数重组断点位于高LTR密度区域,推测这些区域是染色体的候选着丝粒区域(图2G,S4)。此外,使用Hi-C数据(来自相同条件下叶片组织)研究了何首乌与荞麦基因组中同源基因对的三维(3D)染色质A/B compartment。我们发现,何首乌基因组中63%的基因组区域被预测为A compartment,37%的基因组区域被预测为B compartment,而荞麦基因组中分别有54%和46%的基因组区域被预测为A compartment和B compartment(图S5;表S15)。约20.2%和28.6%的何首乌与荞麦之间的共线性区域表现为B到A和A到B compartment的交换(图2E)。KEGG富集分析显示,保守的A/B compartment区域中的基因主要富集在“转录因子”和“运输蛋白”中,而在交换的compartment区域(A到B,B到A)中的基因则主要富集在“亚油酸代谢”和“转录因子”中(图S6)。这些结果表明,何首乌和荞麦的基因组分化后,染色质状态的交换可能改变了参与活性成分生物合成的基因的调控和表达(Oudelaar & Higgs,2021;Song等,2023)。我们还识别出60个大片段(>1 Mb)在何首乌和荞麦基因组之间的倒位。KEGG富集分析显示,接近断点区域的基因在何首乌中显著富集于“植物-病原相互作用”、“细胞色素P450”、“吲哚生物碱生物合成”和“各种植物次级代谢物生物合成”等功能类别中(表S16)。综合而言,基因组进化分析表明,尽管两物种的整体基因组展现了高度的共线性,但在分化后,仍存在显著的结构变异和重排。
3.3 显著扩展的基因家族对何首乌的抗逆性和芪类化合物及蒽醌类化合物生物合成的贡献
基因家族聚类分析显示,何首乌中扩展的基因家族为2623个,特有的基因家族为973个(图2A)。功能富集分析显示,这些基因家族主要富集在“转录因子”、“细胞色素P450”、“植物-病原相互作用”和“异黄酮生物合成”等功能类别中(表S17,S18),这些功能类别此前已被报告与抗逆性和药用活性成分的生物合成相关(Singh等,2021)。在扩展的基因家族中,我们分别在何首乌、荞麦、甜菜(B. vulgaris)和拟南芥(A. thaliana)中鉴定出了221个、158个、137个和154个NBS编码蛋白,115个、68个、56个和75个RLP(受体样蛋白),以及607个、576个、382个和517个RLK(受体样激酶)基因(表S19),并发现何首乌中的这三类抗性基因明显多于其他物种(图S7–S9)。对不同类型重复基因的进一步分析揭示,最近发生的全基因组重复事件,特别是在蓼科植物中发生的事件,对这些基因家族的扩展起到了重大作用(图3A–C,S10;表S20),在何首乌中,81.9%的转录因子和94.1%的抗性基因为全基因组重复基因(图3D)。具体而言,B3转录因子基因家族在何首乌中显著扩展(142个),相比之下,荞麦为97个,甜菜为58个,拟南芥为88个(图S11)。因此,最近发生的全基因组重复事件可能对与抗逆性和活性成分生物合成相关的基因家族扩展起到了重要作用,并进一步帮助何首乌更好地应对生物和非生物胁迫。
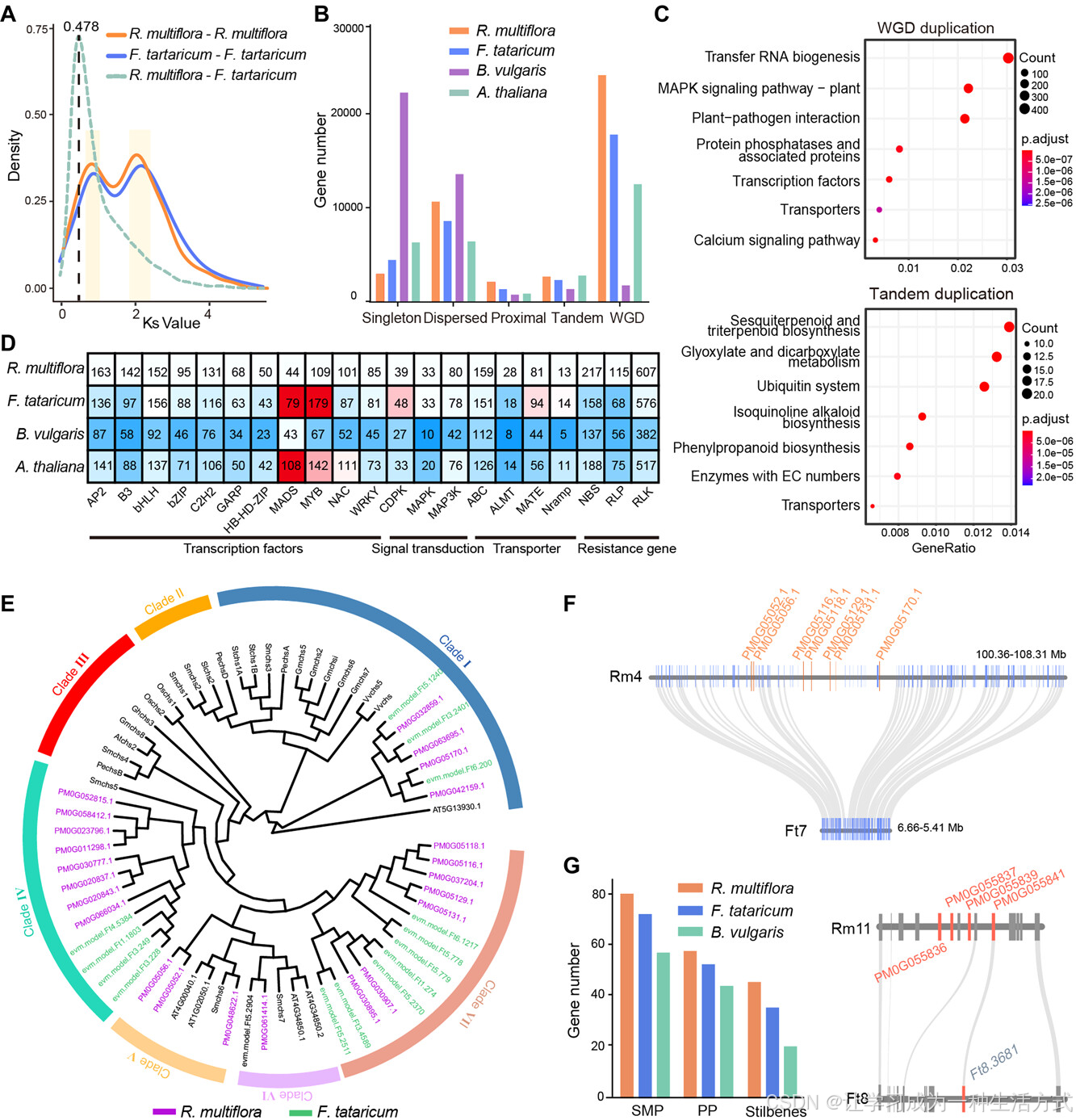
不同类型的重复基因在代谢物生物合成和抗逆性中的作用
A,基因组内同源基因的同义替代(Ks)分布频率。 B,何首乌(R. multiflora)与其他三种物种中重复基因的不同来源分类。 C,分别对来自WGD重复和串联重复的重复基因进行的KEGG富集分析。 D,分析了与抗逆性相关的基因家族在何首乌(R. multiflora)、荞麦(F. tataricum)、甜菜(B. vulgaris)和拟南芥(A. thaliana)中的拷贝数,结果表明,抗逆性相关基因主要来源于WGD重复。 E,基于11种植物物种对何首乌中显著扩展的CHS基因家族的系统发育推测。分支根据右下方的物种颜色方案进行着色。 F,何首乌(Rm)与荞麦(Ft)中的CHS基因微共线性分析。 G,何首乌、荞麦和甜菜中已报告参与芪类化合物和蒽醌类化合物生物合成的基因数量。植物中蒽醌的生物合成主要包括两条途径:多酮途径(PP)和木香酸与甲瓦龙酸/甲基-D-赤藓糖4-磷酸途径(SMP)。何首乌(Rm)与荞麦(Ft)之间O-甲基转移酶基因的微共线性分析。
已报告的Chalcone合成酶(CHS)基因主要参与如黄酮类、芪类和蒽醌类等特种代谢物的生物合成(Zhang, L等,2017;Xia等,2018),并且与荞麦(15个)、甜菜(12个)和拟南芥(5个)相比,何首乌的CHS基因家族显著扩展(23个)(图3E)。CHS基因家族可进一步分为七个亚家族,在七种植物物种中,何首乌的成员主要分布在五个亚家族中,包括Clade I和Clade IV~VII。我们发现,何首乌中Clade IV和Clade V的CHS基因在与其他物种相比时特有的扩展(图3E)。有趣的是,七个CHS基因在何首乌中以串联重复的形式特定分布于第4染色体上,而在荞麦的相应共线性区域中没有发现CHS基因(图3F)。何首乌中显著扩展的CHS基因家族或许部分解释了为何何首乌富含芪类化合物和蒽醌类化合物。
有研究表明,植物中蒽醌类化合物的生物合成可能与多酮途径(PP)和木香酸与甲瓦龙酸/甲基-D-赤藓糖4-磷酸途径(SMP)相关(Kang, S-H等,2021)。通过同源搜索和功能注释,我们在何首乌中鉴定了21个关键基因家族和80个基因,涉及木香酸与甲瓦龙酸/甲基-D-赤藓糖4-磷酸途径,另外三大基因家族和55个基因涉及多酮途径。与荞麦(72和51)和甜菜(57和44)相比,何首乌中的这两条假定的蒽醌合成途径的基因家族(80和55)显著扩展(图3G)。例如,三种O-甲基转移酶基因(催化形成physcion的酶类,Parajuli等,2018)特定插入到了何首乌第11染色体上,而在荞麦中并未出现(图3G)。此外,我们还鉴定了四个基因家族和40个基因,参与了芪类化合物生物合成途径,何首乌中这些基因(40个)显著多于荞麦(34个)和甜菜(20个)(图3G)。我们发现,这些基因中63.69%是由最近的WGD事件产生的重复基因,10.19%是由串联重复产生的,这表明最近的WGD事件在何首乌中对与蒽醌和芪类化合物生物合成相关的基因家族的演化产生了重要影响,极大地促进了其对环境胁迫的抵抗能力。
3.4 根系发育的代谢物谱和转录组学分析
先前的研究表明,何首乌的活性成分主要集中在根部(Zhao等,2021)。为了深入了解何首乌的次生代谢物生物合成及其相关的调控路径,我们分别获得了不同生长年限(1年生[OY]、3年生[TY]、5年生[FY]、10年生[TEY])根部的转录组学和代谢组学数据(图4A)。在四个不同生长年限的根部共检测到783种次生代谢物(图S12;表S21),这些代谢物的层次聚类分析显示,次生代谢物的总含量在FY根部最高(图4A,S13)。与其他次生代谢物相比,各年限根部芪类化合物的平均含量和总含量最高,而蒽醌类化合物的平均和总含量在这四个根部中变化最大(图4B,S14)。总体而言,随着根部生长,蒽醌类化合物和芪类化合物的丰度逐渐积累,但蒽醌类化合物的含量在第五年生长时达峰值,而芪类化合物的含量在第十年最高。在芪类化合物中,THSG、THSG、2,3,5,4′-四羟基stilbene-2-O-(2′-没食子酸酰基)-D-glucoside和2,3,5,4′-四羟基stilbene-2-O-(2′-没食子酸酰基)-D-glucoside分别在OY、TY、FY和TEY根部含量最高(表S22)。对于蒽醌类化合物,Physcion-8-O-(6-乙酰基)-glucoside在OY、TY和FY根部含量最高,而6-甲基芦荟蒽醌在TEY根部含量最高(表S23)。此外,我们发现,何首乌中的芪类化合物和蒽醌类化合物主要以糖苷形式存在,如THSG、physcion 8-O-glucoside、emodin 8-O-glucoside和emodin 6-O-glucoside。简而言之,代谢组学分析显示,何首乌根部富含芪类化合物和蒽醌类化合物,而这四个不同生长年限的根部在这些代谢物的种类和含量上存在显著差异。这些结果表明,动态变化的生物活性化合物可能有助于何首乌强大的环境适应性。
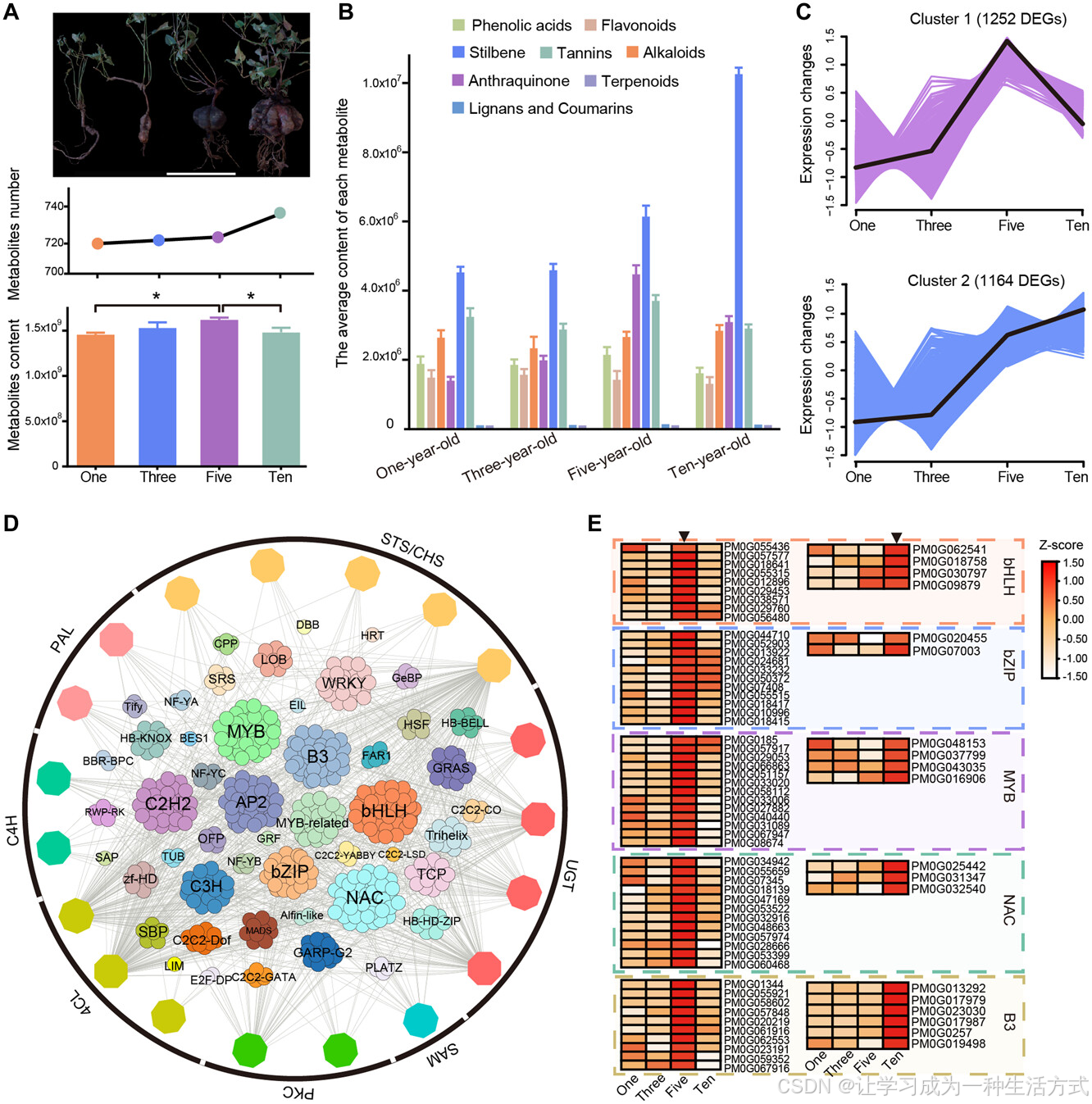
A,何首乌1年、3年、5年和10年生根的形态,以及不同生长年限的根中次生代谢物的总类型和相对含量,具有统计学显著性(*P < 0.05,Student's t检验)。比例尺 = 15 cm。 B,不同生长年限根中不同类别次生代谢物的相对含量。 C,四个不同生长年限的根部中差异表达基因(DEGs)的层次聚类,包括集群1(5039个基因)和集群2(2894个基因),它们分别与芪类化合物和蒽醌类化合物的积累高度相关。 D,芪类化合物和蒽醌类化合物生物合成结构基因与转录因子之间的共表达网络。填充颜色的六边形表示与生物合成相关的结构基因。 E,不同生长年限根中五个转录因子家族的表达,且大多数转录因子在5年生和10年生根中高度表达。条形图表示每个基因的表达水平(z得分)。
转录组学分析揭示,在不同生长年限的根部之间共识别出15,505个差异表达基因(DEGs)(图S15–S17;表S24、25)。功能富集分析显示,这些DEGs主要富集在“细胞色素P450”和“运输蛋白”类别,可能参与何首乌中活性成分的合成、运输和储存,以及抗逆反应(表S26)。在这些DEGs中,鉴定出1021个转录因子,其中排名前七的转录因子家族分别为AP2(95个)、bHLH(78个)、C2H2(63个)、bZIP(59个)、NAC(59个)、MYB(58个)和WRKY(57个),表明它们在根系生长过程中可能在调控活性成分积累中发挥潜在作用(表S27)。为了阐明不同生长年限根系间的基因表达模式,我们通过k-means聚类分析将所有DEGs划分为12个集群(图S18)。我们观察到,集群1和集群2中的基因表达模式分别与芪类化合物和蒽醌类化合物的积累高度相关(图4C)。此外,基于基因表达水平与活性成分含量的相关性,我们通过WGCNA(图S19、S20;表S28)获得了共表达模块。其中,两个共表达模块(青绿色模块和棕色模块)与蒽醌类化合物和芪类化合物的生物合成显著相关(图S21、S22)。这些模块可能作为未来关于蒽醌类化合物和芪类化合物生物合成的遗传分析的优选候选模块(图S21、S22)。转录组与代谢物的相关分析表明,许多转录因子的表达模式与参与芪类化合物和蒽醌类化合物生物合成的基因家族高度相关,这些转录因子,如NAC、bHLH、MYB、B3和AP2,可能是这些酶基因的潜在直接调控因子(图4D;表S29)。此外,我们还发现,大多数转录因子在5年生根中高度表达,这与蒽醌类化合物含量的变化趋势一致,而一些转录因子在10年生根中高度表达,这与芪类化合物含量的变化趋势一致(图4E,S23、S24;表S30)。这些转录因子的高表达可能促进了天然产物含量的增加,这对于植物增强对生物和非生物胁迫的抵抗能力至关重要(Isah,2019;Li等,2023)。
3.5 何首乌蒽醌类化合物生物合成关键基因的鉴定
何首乌中的丰富蒽醌类化合物已经临床证明是主要的生物活性化合物,包括大黄素、physcion及其衍生物(Zhao等,2021)。结合四种不同生长年限的根及三种不同组织的转录组学和代谢组学分析,我们推测并提出了何首乌大黄素及其衍生物的生物合成途径。共鉴定出58个候选基因,属于四个参与大黄素及其衍生物生物合成的基因家族,包括18个CHS基因、16个PKC基因、16个OMT基因和8个UGT73基因(图5B,5C;表S31)。有趣的是,这些基因家族大多扩展了,何首乌中CHS同源基因的数量(18个)相较于荞麦(12个)、甜菜(12个)和拟南芥(5个)显著增加。转录组分析显示,65.5%的候选基因在七个样本中表达,并且每个基因家族至少有一个基因高度表达。其中,四个CHS基因(PM0G032859、PM0G042159、PM0G05170、PM0G063659)和四个PKC基因(PM0G030326、PM0G031295、PM0G039312、PM0G032418)在所有不同生长年限的根中始终高度表达,这与我们代谢组学分析中所有根系组织中大黄素的高含量一致(图5B)。此外,代谢组学分析显示,physcion的含量在五年生根中达到峰值。大多数OMT基因在不同生长年限的根中始终表达,但三个基因(如PM0G058422、PM0G057511和PM0G055836)在FY根中特异性表达,表明这三个基因可能是FY根中physcion显著积累和最高含量的原因(图5B)。研究表明,FtUGT73BE5催化了emodin 6-O-glucoside的形成。我们在何首乌基因组中鉴定了126个UGT基因。根据系统发育树,RmUGT基因簇分为13个组(A–B,D–H,J–N,和P)(图5G;表S32)。其中,属于D组的RmUGT基因主要在何首乌根组织中高度表达,且两个基因(即PM0G031940和PM0G054254)在5年生根中表现出最高的表达(图5B,5G),这些基因可能催化大黄素及其衍生物的糖苷化反应(表S32)。这些RmUGT73基因的转录组学分析结果与代谢组学结果高度一致,表明physcion 8-O-glucoside和emodin 6-O-glucoside的含量在5年生根中最高。因此,CHS、PKC、OMT和UGT73基因的拷贝数增加和表达模式对何首乌大黄素及其衍生物合成的调控至关重要,可能促进了其对环境的适应能力。
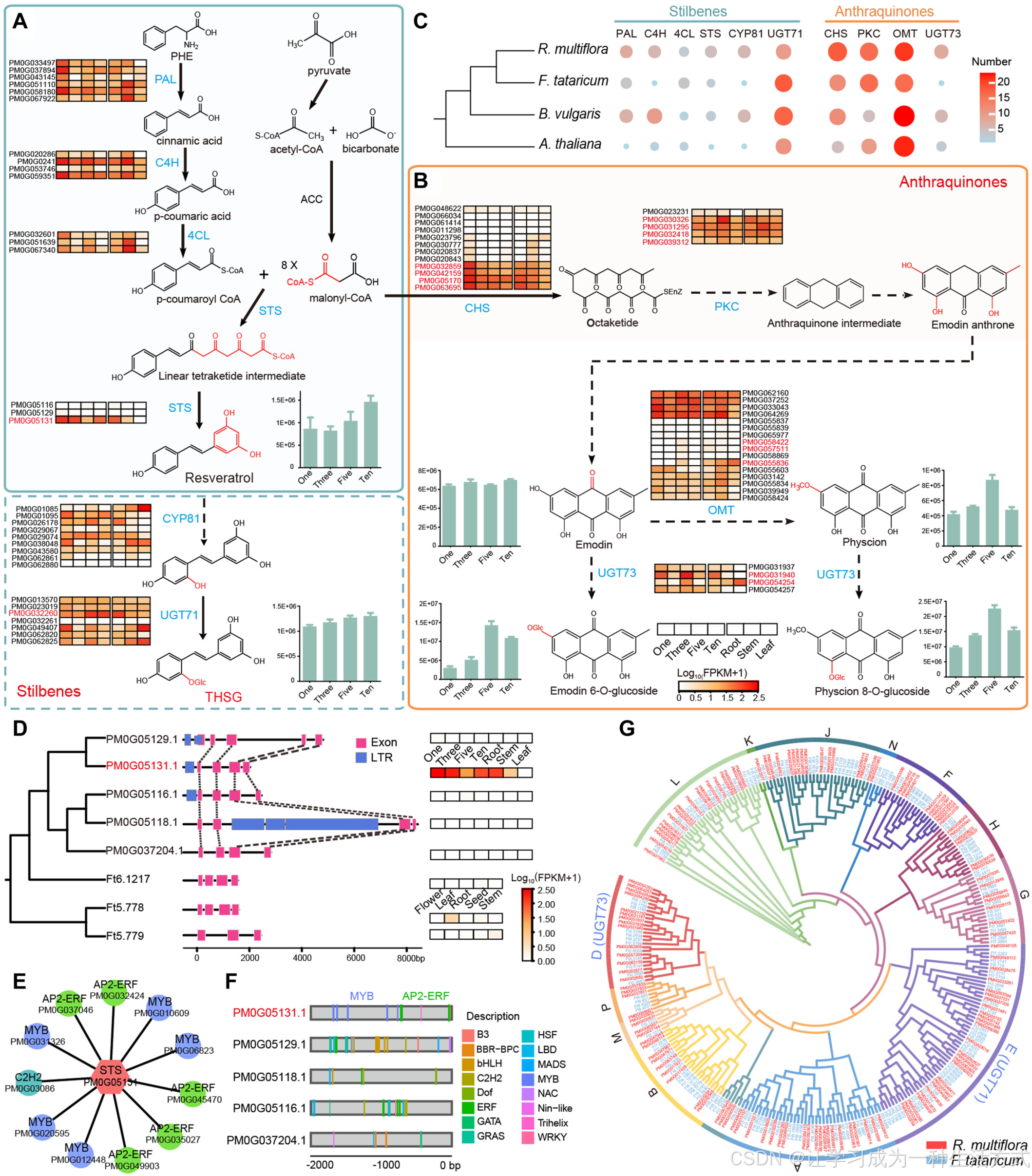
A 和 B,推测的芪类化合物和蒽醌类化合物生物合成途径及相关关键候选基因的表达。虚线表示多个步骤。热图展示了不同样本中基因表达水平。蓝色字体代表参与催化步骤的酶的缩写。柱状图展示了不同生长年限根中代谢物相对含量的变化。纵轴代表代谢物丰度。 C,参与芪类化合物和蒽醌类化合物生物合成途径的不同基因家族的拷贝数。 D,何首乌(R. multiflora)和荞麦(F. tataricum)中STS基因的系统发育、基因结构和表达谱。 E,11个被识别为STS基因(PM0G05131)直接调控因子的转录因子(五个MYB、五个AP2-ERF和一个C2H2)。 F,检测到的位于何首乌五个STS基因2kb上游序列中的转录因子结合位点。 G,何首乌基因组中UDP-糖苷转移酶(UGT)基因家族的系统发育。
3.6 THSG生物合成中的关键基因的鉴定与分析
THSG是何首乌中最重要的活性成分之一,并根据《中国药典》被作为何首乌鉴定的主要理化标志物(Lv等,2007;Lin等,2015)。我们的代谢组学分析显示,芪类化合物是何首乌中最丰富的代谢物,THSG在这些芪类代谢物中最为普遍。虽然如白藜芦醇的生物合成途径已经有较为清晰的描述,主要来源于苯丙烷途径,但THSG的生物合成途径仍然不明,尤其是从白藜芦醇转化为THSG的途径。
白藜芦醇是通过一个已知的生物合成途径产生的,该途径涉及四种主要酶:PAL、C4H、4CL和STS。与白藜芦醇相比,THSG在其C2位置具有额外的O-糖苷基。已有报道指出,CYP81能够催化C2位置的羟基化(Liu等,2003),而UGT71A16能够催化类黄酮在2′-O位置的糖苷化(Gosch等,2010)。因此,我们推测CYP81的同源基因可能参与催化白藜芦醇的羟基化,产生THSG aglycone,而UGT71A16的同源基因则可能参与催化THSG aglycone的糖苷化反应,从而在何首乌中合成THSG。
我们共鉴定了40个候选基因,编码六种关键酶,参与THSG生物合成途径,包括8个PAL,6个C4H,3个4CL,5个STS,8个CYP81和10个UGT71基因(图5A,5C,S25;表S33)。值得注意的是,我们发现THSG生物合成途径中关键中间代谢物的含量变化与相关酶基因在不同生长年限组织中的表达变化始终一致。我们发现,STS基因家族在何首乌中(五个基因)较荞麦(三个基因)、甜菜(三个基因)和拟南芥(零个基因)更多。特别是转录组结果显示,何首乌中的一个STS基因(PM0G05131)在根部的表达水平显著高于其他组织,并且在不同生长年限的根部中持续高度表达(图5A)。代谢组分析显示,根部中的白藜芦醇含量随着根部的生长而积累。因此,我们推测PM0G05131在根部的显著高表达可能是导致何首乌根部持续合成白藜芦醇的原因。系统发育与比较分析显示,PM0G05131的基因结构与其他STS基因保守(图5D,S26)。然而,从时间排序的基因共表达网络(TO-GCN)推断的共表达网络显示,PM0G05131在涉及五个乙烯响应因子(ERFs)、五个MYB和一个C2H2的潜在调控网络关系中是核心节点(图5E)。有趣的是,转录因子(TFs)结合位点的分析揭示,PM0G05131的2kb上游序列中的潜在调控元件主要是ERFs和MYB TFs,这与其他STS基因显著不同(图5F)。因此,我们推测PM0G05131基因可能受MYB和ERFs作为调控因子的调控,这与先前的研究结果一致,即ERF通过与MYB相互作用调控葡萄中的芪类化合物合成(Wang & Wang,2019)。此外,大多数何首乌中的RmCYP81和RmUGT71基因在四个不同生长年限的根中始终高度表达,这可能直接促使THSG在何首乌中的持续积累。因此,STS、CYP81和UGT71基因的拷贝数增加和表达模式对于何首乌中THSG的生物合成至关重要,这可能部分解释了何首乌为何富含THSG。




