目录
1.最快简介
2.Samtools 安装
3.SAM vs BAM
4.Sam/Bam文件格式
(1)Header section
(2)SAM/BAM 数据行
QNAME:
FLAG
RNAME
POS
MAPQ
CIGAR
1.最快简介
SAM(Sequence Alignment/Map)和 BAM(Binary Alignment/Map)文件格式由 The Sequence Alignment/Map format specification 组织创建,旨在为基因组学和生物信息学提供一种标准化的方式来表示序列比对信息。
SAM 和 BAM 文件广泛应用于基因组测序数据的存储和分析,尤其是在高通量测序技术(如NGS或者RNA-seq)中。它们用于存储测序读段与参考基因组的比对信息,方便后续的变异检测、基因表达分析和其他生物学研究。
学习 SAM 和 BAM 文件格式非常重要,随着生物信息学的快速发展,相关工具和软件(如 SAMtools 和 GATK)都基于这些文件格式进行操作,了解它们是深入该领域的基础。
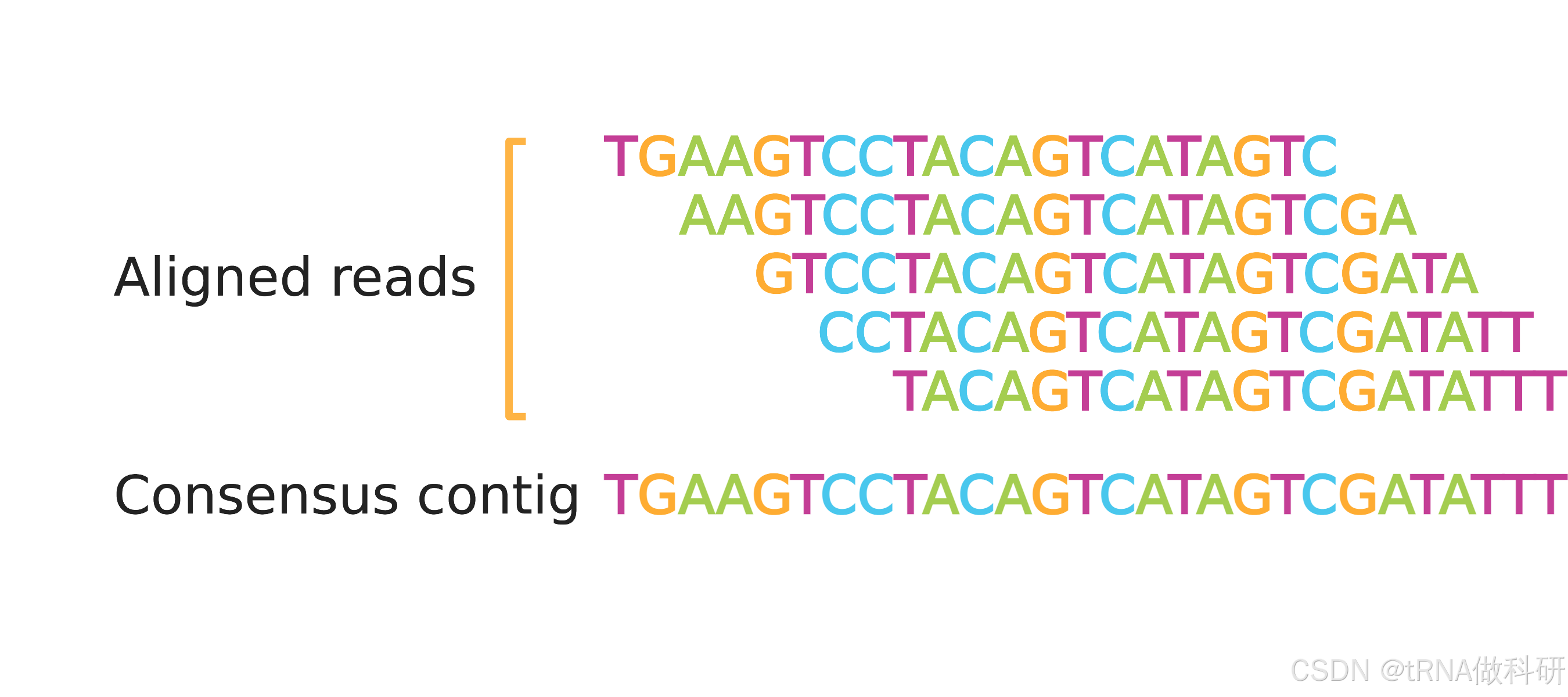
2.Samtools 安装
我们通过conda进行工具管理与包安装:
#如果需要新创建环境
conda create -n samtools
conda activate samtools
#在新环境或当前环境安装
conda install bioconda::samtools
验证安装:
samtools --help
3.SAM vs BAM
SAM 文件是制表符分隔的文本文件,其中包含每个单独读取的信息及其与基因组的比对信息。
BAM 文件只是一个压缩的 SAM 文件。该文件以 Header(可选)开头。
Header用于描述数据源、参考序列、对齐方法等,这将根据所使用的对准器而变化。
Header后面是对齐部分。每一行对应于单个读取的对齐信息。每条基准都有 11 个必填字段,用于基本映射信息,以及数量可变的其他字段,用于对准器特定信息。
4.Sam/Bam文件格式
(1)Header section
标题部分位于对齐部分之前。如果存在,它可以包含任何信息,但主要是meta信息,如 SAM 版本、生成文件的命令或有关对齐中使用的参考信息。
标题行以@字符开头。
简单例如下:
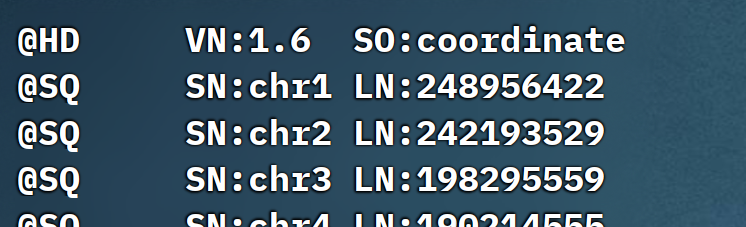
那这些标识的含义是什么呢,可以详细看下面的表格
一定记住不是每个参数都会出现在文件中
首先是HD开头的行:记录一些总体信息

然后是SQ开头的行:记录比对

RG开头的行:用于排序
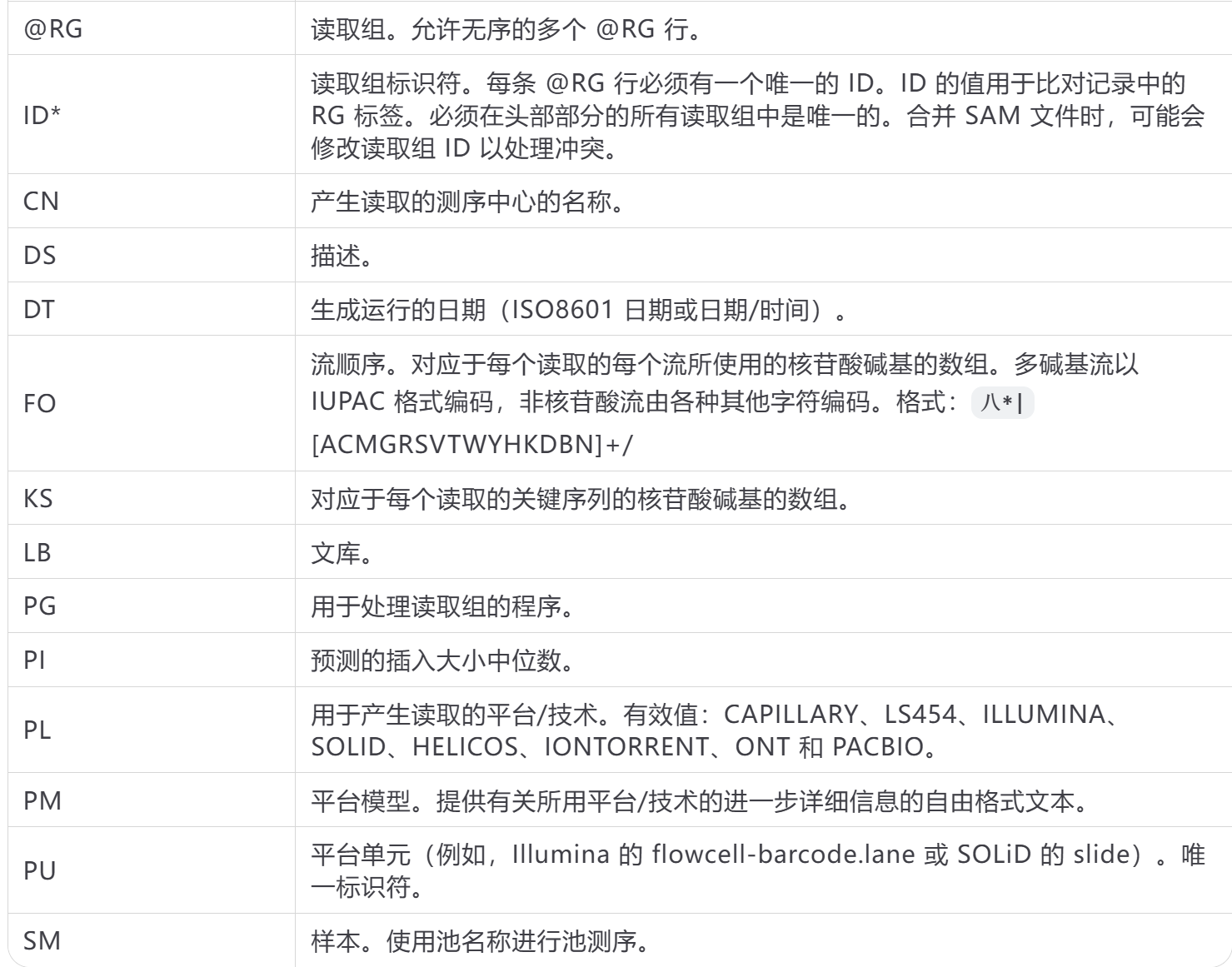
最后是PG行:记录程序运行情况
CO行:记录一些其他的注释

我们用另一个例子做说明:

第一行以@SQ 开头,表示它正在识别一个参考序列 contig。
“SN”标签表示 contig 名称(chr14),而“LN”标签表示 contig 长度(107349540 个碱基)。
在这个例子中,只有一个参考序列 contig 是染色体 14,但大多数参考基因组将包含几个 contigs 和几条@SQ 行以匹配。
第二行以@PG 开头,表示它描述了用于生成 SAM 文件的程序(进行了序列比对)。
如果有多个 SAM 文件合并,可能会出现多个@PG 行,尽管通常只有一个。“ID”标签是此程序在此 SAM 文件中的唯一标识符(如果使用了多个 BWA 版本,则必须修改 ID 值以避免冲突)。“PN”标识了使用的程序名称(在文件中不需要唯一),“VN”标签标识了程序的精确版本。“CL”标签提供了执行对齐所使用的命令的副本。
(2)SAM/BAM 数据行
BAM 或 SAM 文件中的基本数据单元是 BAM 或 SAM 行,它包含单个 NGS read的读取和对齐数据。
BAM 和 SAM 文件中数据的编码略有不同,但信息本身在这两个文件之间不会改变。
我们在文件中看到的例如下:
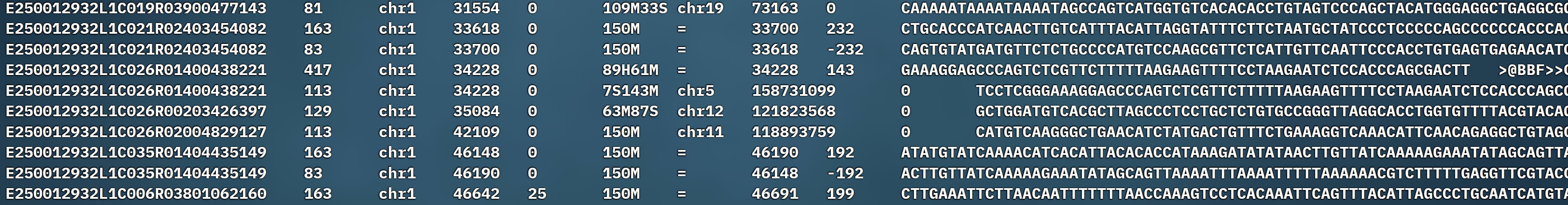
每行包含 11 个必填字段:
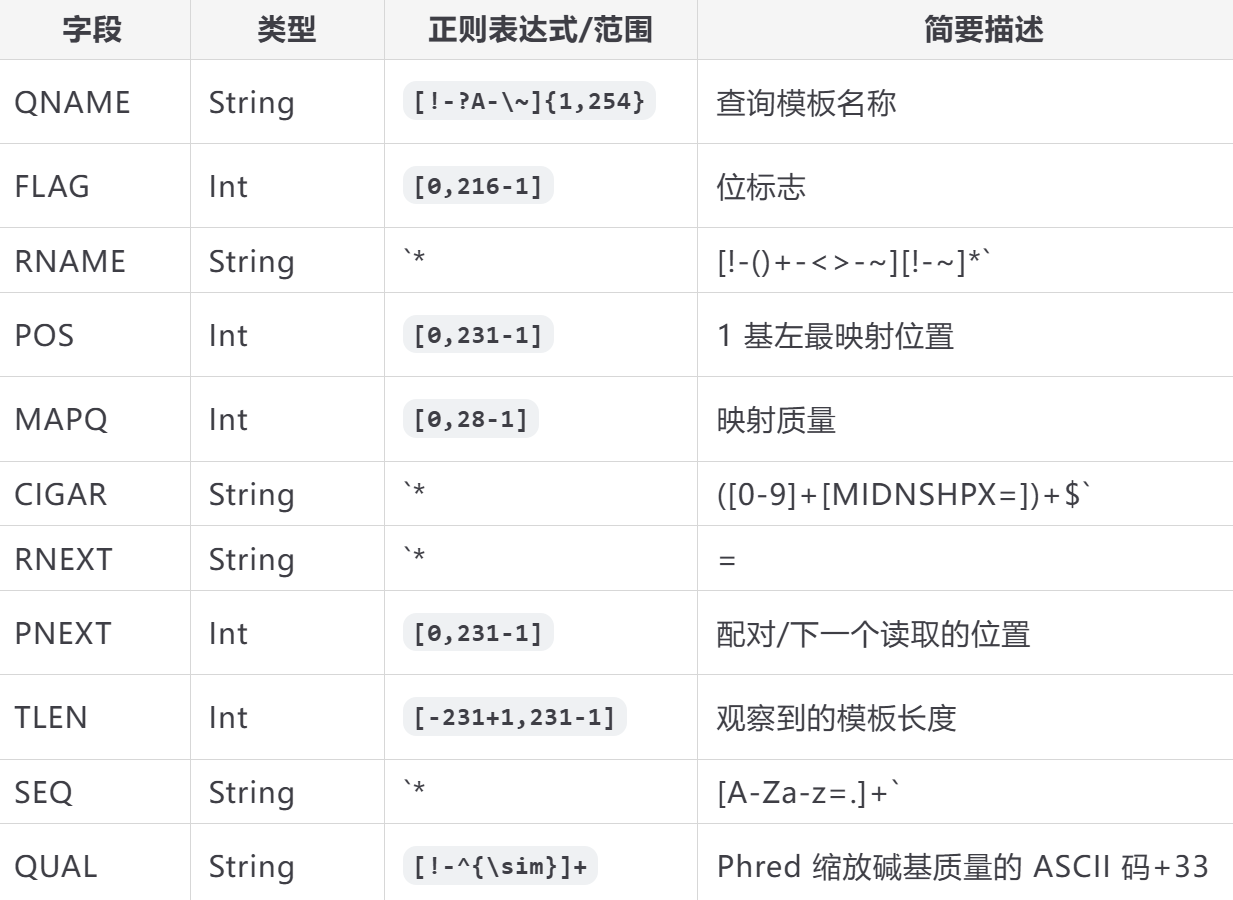
这就很难理解了,我们以例子的形式看其中比较重要和难理解的内容:
QNAME:
QNAME 是文件中读取的唯一标识符,可以用来识别任何单个读取。例外情况是配对末端读取(paired-end reads)将具有相同的 QNAME,因为它们来自同一 DNA 片段,并且必须通过它们的方向(可以从 FLAG 值确定)来区分。
FLAG
是一个用于表示二进制的数,该数以数字表示不同的真/假判断,这些数值与读取的对齐情况有关。
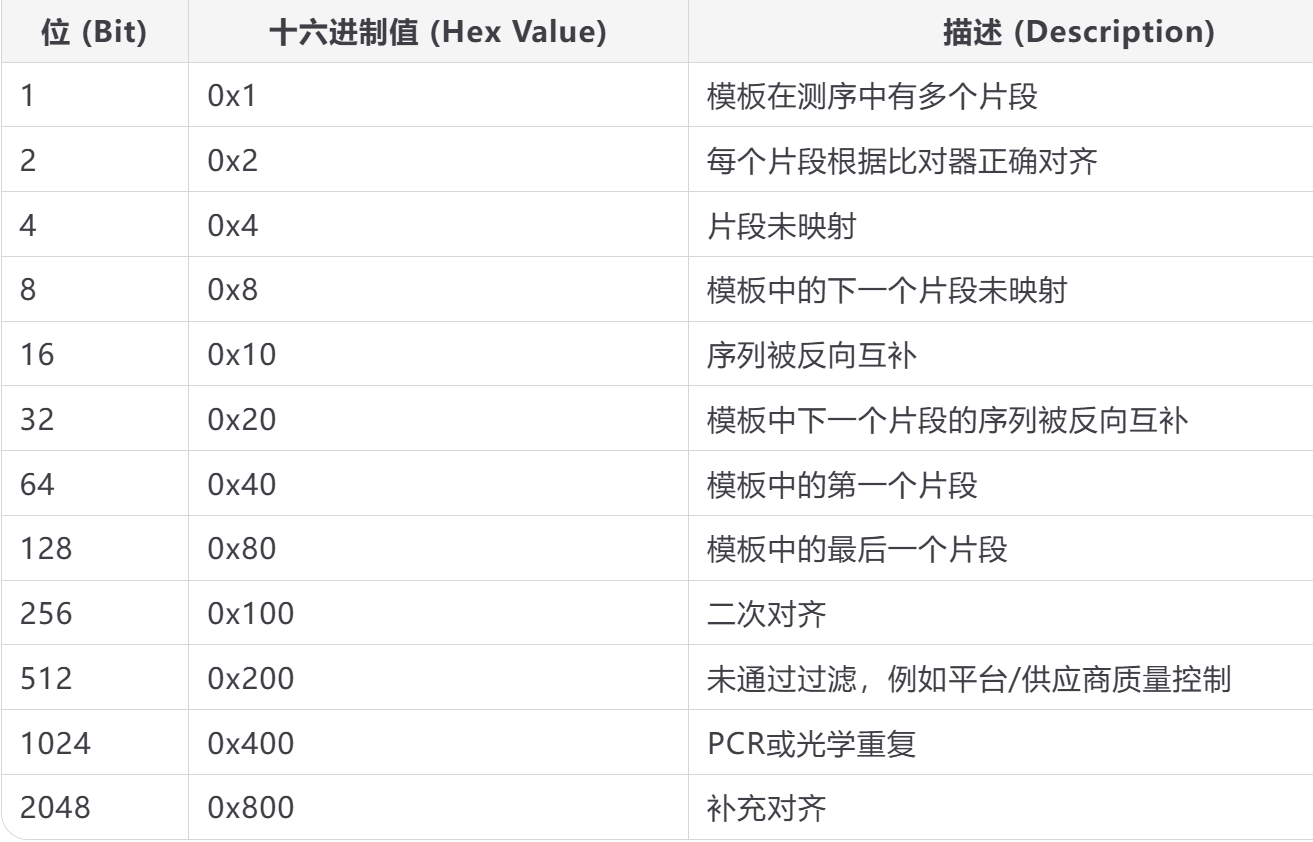
但是大部分时候FLAG不是其中的数字,而是一个复合计算数字。
例如,一个常见的正确 FLAG 值是 99,它由以下组成:
64 + 32 + 2 + 1,解释为:
读取是成对中的第一个(64)
该读取的配对端映射在反向方向(32)
读取是正确对齐的一对的一部分(2)
读取是配对的(1)
FLAG 值的一个有趣特性,由于 1 表示读取是配对的,因此pair end比对应该总是产生奇数 FLAG 值,奇数 FLAG 值总是表示配对端比对。
RNAME
标识了参考基因组中哪个 contig 与读取的序列进行了对齐。此值应存在于一个@SQ 行中的标题中。
POS
表示读取中第一个匹配碱基的左侧映射位置(有关匹配构成的信息,请参阅 CIGAR)。
位置值是基-1,这意味着参考序列的第一个字母被计为位置 1(其他系统可能将此位置计为 0)。通常,参考名称和位置统称为基因组坐标(the genomic coordinates)。
MAPQ
映射质量是一个 Phred 缩放的置信度分数,表示序列被正确或错误映射的可能性。此字段中的 255 值表示未给出概率,被视为占位符值。
不同的算法报告方式不同,但无论如何,数值越大,对齐(通常)越好。
CIGAR
表示由插入或删除的碱基(或其他导致不连续的原因)引起的比对中的连续性或不连续性。一个典型的 CIGAR 可能看起来像 5M2D5M,以说明序列的哪些部分对齐,哪些不对齐,以及介于两者之间的所有内容。

在 5M2D5M 的上述情况中,有 5 个碱基同时存在于读取和参考中,2 个碱基在读取中缺失但在参考中存在,然后又有另外 5 个碱基同时存在于两者中(请注意,在这 5 个匹配的碱基片段中,可能存在实际的序列不匹配,即参考和读取中的碱基不同,因为匹配仅表示存在)。
剪切操作 S 和 H 需要进一步解释:
软剪辑: 更为常见,指的是对齐不始于读取的末端,而是在其中某个位置开始的情况。因此,对齐两侧的读取部分在 CIGAR 字符串中被标记为 '软剪辑',这样其他软件就知道不要使用它,例如在变异调用中。
硬剪辑: 用于报告辅助对齐,即当读取被分割以创建多个对齐时,其余对齐到其他位置的读取部分被硬剪辑,即从记录的 SEQ 中省略。
我们通过几个例子看看理解程度:
例1:
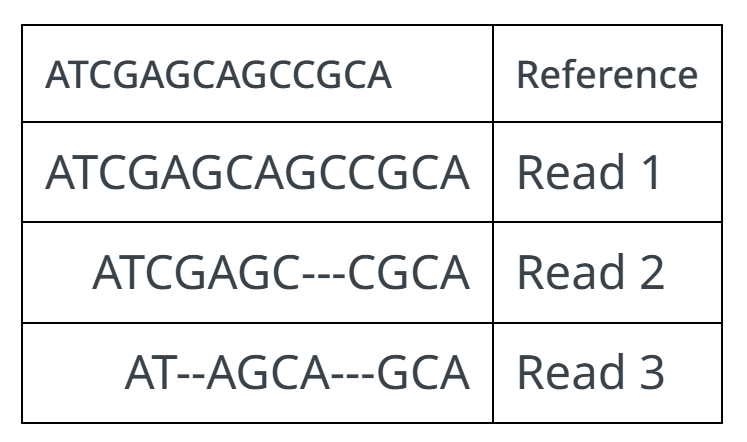
可以分别表示为:
14M:此序列完全匹配
7M3D4M:读取中缺少 3 个碱基
2M2D4M3D3M:由 4 个碱基分隔的 2 碱基和 3 碱基缺失
例2:

可以分别表示为:
14M:此序列完全匹配
6M2I8M:读取中存在 2 个碱基,但参考中不存在
例3:

可以分别表示为:
14M:此序列完全匹配
14M:中间的长串“T”错配仍然被视为匹配位置
以上就是全部的内容
下一篇将更新具体的SAM和BAM操作

)
感知机)


